Voir la traduction automatique
Ceci est une traduction automatique. Pour voir le texte original en anglais cliquez ici
#Actualités du secteur
{{{sourceTextContent.title}}}
Présentation de néoantigènes tumoraux
{{{sourceTextContent.subTitle}}}
Présentation de néoantigènes tumoraux
{{{sourceTextContent.description}}}
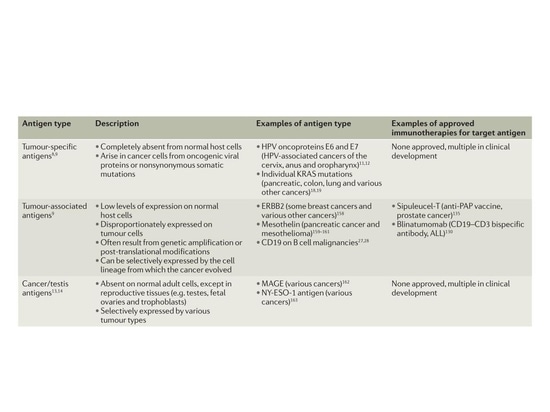
Classification des antigènes tumoraux
Les dernières avancées en matière de séquençage du génome indiquent qu'au cours de son initiation et de son développement, le cancer acquiert des dizaines de milliers de mutations cellulaires somatiques différentes. La plupart de ces mutations ne confèrent pas d'avantages inhérents à la croissance (mutations passagères) et sont souvent le résultat d'une instabilité génomique au sein de la tumeur.
Un petit sous-ensemble de mutations cancéreuses perturbe la régulation cellulaire normale et contribue à la croissance du cancer et à la résistance aux thérapies ciblées (mutations pilotes). Jusqu'à présent, environ 140 gènes ont été identifiés comme pouvant être à l'origine de la tumorigenèse. Cependant, les mutations "driver" et "passenger" peuvent toutes deux modifier la séquence codante des acides aminés, ce que l'on appelle collectivement les mutations synonymes, et conduire à des protéines mutées qui ne sont pas exprimées dans les cellules normales. Ces séquences protéiques aberrantes sont transformées en peptides courts et se lient au complexe majeur d'histocompatibilité (CMH ; également connu sous le nom d'antigène leucocytaire humain HLA chez l'homme), ce qui les rend reconnaissables par les cellules T en tant qu'antigènes étrangers [1].
En raison de leur expression sélective dans les tumeurs, les antigènes spécifiques des tumeurs (TSA) générés par des mutations synonymes et d'autres altérations génétiques sont appelés néoantigènes. Dans les sous-groupes de tumeurs humaines d'étiologie virale, tels que le carcinome à cellules de Merkel (MCC) associé au polyomavirus à cellules de Merkel (MCPyV) et les cancers du col de l'utérus, de la bouche et d'autres sites spécifiques associés au papillomavirus humain (HPV), les protéines codées par le cadre de lecture ouvert viral représentent un autre type de néoantigène. Outre les TSA, il existe deux grandes classes d'antigènes tumoraux. Les antigènes associés aux tumeurs (TAA) sont surexprimés dans les cellules malignes mais sont également exprimés à de faibles niveaux dans les cellules normales. Les antigènes du cancer/testicule (CTA) sont exprimés par divers types de tumeurs et de tissus reproducteurs (par exemple, les testicules, les ovaires fœtaux et les couches nourricières), mais leur expression est limitée dans d'autres tissus adultes normaux et ils sont généralement absents des cellules reproductrices normales car ces tissus n'expriment pas les molécules de classe du CMH. Les néoantigènes peuvent apparaître au niveau génomique par des variations de nucléotides simples (SNV), des insertions de bases et des fusions de gènes, au niveau transcriptionnel par l'épissage sélectif, la polyadénylation (pA), l'édition de l'ARN et les régions dites non codantes, et au niveau protéique par une traduction perturbée et des modifications post-traductionnelles.
▲Classification des antigènes tumoraux[1]
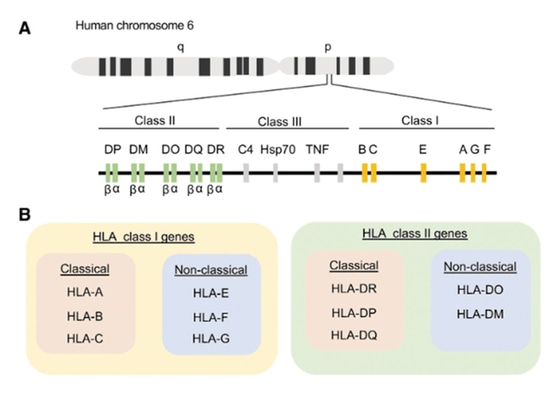
Classification des HLA
L'activation des cellules T dépend de la reconnaissance simultanée de fragments de peptides étrangers et de molécules du CMH du soi, un phénomène connu sous le nom de restriction du CMH. Les cellules T CD8+ sont restreintes par le CMH-I, tandis que les cellules T CD4+ sont restreintes par le CMH-II. Le complexe majeur d'histocompatibilité (CMH), appelé antigène leucocytaire humain (HLA) chez l'homme, est codé sur le chromosome 6 du génome humain. Il s'agit d'un complexe de gènes hautement polymorphes qui codent pour des molécules de surface cellulaire spécialisées dans la présentation et la reconnaissance de peptides du soi et du non-soi. Le HLA est divisé en trois classes en fonction de sa fonction et de sa structure : HLA-I, HLA-II et HLA-III. Les molécules de classe HLA sont exprimées à la surface des cellules nucléées, à l'exception des cellules germinales et de certaines cellules neuronales. Contrairement aux molécules de classe HLA-I, les molécules de classe HLA-II se trouvent généralement sur les cellules professionnelles présentatrices d'antigènes (APC) telles que les cellules B, les macrophages, les cellules dendritiques, les cellules de Langerhans, l'épithélium thymique et les cellules T activées (plutôt qu'au repos). La structure et la fonction des molécules de classe HLA-III ne sont pas bien comprises, mais on sait qu'elles participent au processus inflammatoire sans se lier directement aux antigènes.
Le système HLA se subdivise en gènes classiques et non classiques. Les gènes HLA-I classiques comprennent HLA-A, HLA-B et HLA-C, tandis que les allèles non classiques comprennent HLA-E, HLA-F et HLA-G. Les gènes HLA-II classiques comprennent HLA-DR, HLA-DP et HLA-DQ, tandis que les allèles non classiques comprennent HLA-DO et HLA-DM. Les cellules T CD8+ humaines reconnaissent les peptides présentés par les gènes HLA-A et HLA-B classiques et, dans une moindre mesure, par le gène HLA-C. Les cellules T CD4+ humaines reconnaissent les peptides présentés par les gènes HLA-A et HLA-B classiques. Les cellules T CD4+ humaines reconnaissent les peptides présentés par HLA-DR, HLA-DQ et HLA-DP. HLA-I consiste en trois domaines extracellulaires (α1, α2 et α3) liés de manière non covalente à une molécule de β2-microglobuline. HLA-II est un hétérodimère composé d'une chaîne α et d'une chaîne β. Les domaines extracellulaires de HLA forment une fente de liaison à l'antigène, constituée de deux hélices α entourant des feuillets β antiparallèles. Cela crée une plate-forme capable d'accueillir un court segment d'acide aminé (aa) appelé peptide. Ces peptides se fixent au fond du sillon de liaison grâce à des interactions avec des acides aminés spécifiques (appelés résidus d'ancrage). En raison de la structure fermée de la fente de liaison du HLA-I, il se lie généralement à de petits peptides de 8 à 10 acides aminés, tandis que le HLA-II peut se lier à des peptides plus longs, dépassant 11 acides aminés.
▲Classification des gliomes diffus chez l'adulte[2]
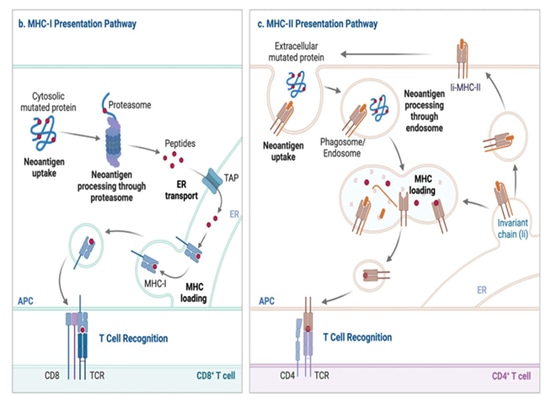
Présentation des néoantigènes
La présentation des protéines du CMH-I provient principalement de peptides à l'intérieur de la cellule. Au cours de l'homéostasie, les protéines cellulaires sont dégradées par les protéasomes, ce qui génère de petits peptides. Les protéasomes sont des complexes multiprotéiques qui dégradent les protéines en petits fragments peptidiques. Lors d'une infection virale, l'action des interférons induit la formation d'un complexe protéasomique alternatif appelé immunoprotéasome, qui favorise la génération de peptides présentés par le CMH-I. Par conséquent, lors de la synthèse des protéines virales, celles-ci sont ciblées pour être dégradées par l'immunoprotéasome, soit sous forme de protéines entièrement repliées, soit sous forme de produits ribosomaux défectueux. Il en résulte la production de petits fragments peptidiques dérivés du virus infectant, qui peuvent être modifiés par des aminopeptidases cytoplasmiques.
Ces fragments peptidiques sont ensuite transportés dans le réticulum endoplasmique (RE) par un mécanisme de transport de protéines connu sous le nom de protéine de transport liée au traitement de l'antigène (TAP). Dans le RE, les peptides subissent une nouvelle modification par l'aminopeptidase 1 du réticulum endoplasmique (ERAP1) s'ils sont trop longs pour se lier au CMH-I après leur translocation par la TAP. Dans le RE, les molécules de CMH-I vides s'associent à des complexes de chargement de peptides (PLC), comprenant des protéines chaperonnes telles que la tapasine et la calnexine. Le PLC maintient le CMH-I vide dans une conformation réceptive aux peptides et, lorsque les peptides sont transportés par la TAP, cette conformation facilite la liaison des peptides. La TAP fait également partie de la PLC. La liaison des peptides stabilise la protéine du CMH-I, la libérant des partenaires de contrôle de qualité du réticulum endoplasmique, et elle est transportée à la surface de la cellule par l'appareil de Golgi.
Ce processus permet aux cellules T CD8+ de rechercher des signes d'infection ou de malignité dans le répertoire des protéines cellulaires. Cependant, dans des sous-ensembles spécifiques de cellules dendritiques, il existe une voie alternative, connue sous le nom de présentation croisée, qui implique l'absorption d'antigènes extracellulaires, leur transfert rétrograde des phagosomes vers le cytoplasme et leur dégradation ultérieure pour le chargement du CMH-I dans les protéasomes et le RE.
▲Les voies de production et de présentation des néoantigènes[3]
L'homme possède plus de 24 000 allèles différents d'antigènes leucocytaires humains de classe I (HLA-I, y compris HLA-A, -B et -C) et de classe II (HLA-DR, HLA-DQ et HLA-DP), ce qui donne lieu à un polymorphisme diversifié. La combinaison de ces allèles contribue à la diversité du polymorphisme. Les allèles HLA du patient déterminent le répertoire des néoantigènes spécifiques de la tumeur, qui sont présentés pour être reconnus par les cellules T. En outre, la perte d'allèles HLA peut entraîner une perte d'identité. En outre, la perte d'hétérozygotie HLA (HLA-LOH), qui se produit dans 40 % des cancers du poumon non à petites cellules, entrave la présentation des néoantigènes, favorisant ainsi l'évasion immunitaire. Par conséquent, l'une des premières étapes cruciales de la prédiction des néoantigènes est l'identification du génotype HLA du patient. Diverses méthodes informatiques peuvent désormais être appliquées aux données de séquençage de nouvelle génération (NGS) pour atteindre cet objectif.
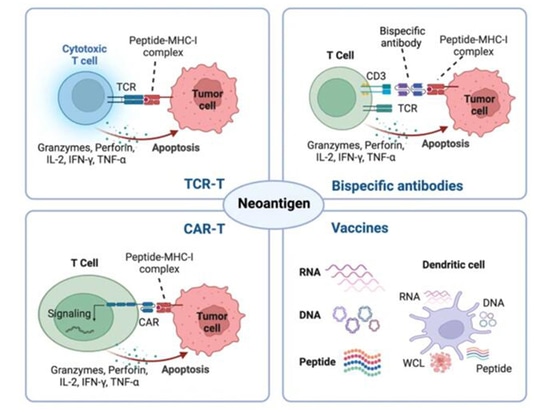
Stratégies thérapeutiques basées sur les néoantigènes
En raison de l'absence de sélection thymique et de tolérance centrale, les néoantigènes spécifiques des tumeurs générés par des altérations génétiques provoquent l'apparition de cellules T à haute affinité. Grâce à leur spécificité tumorale et à leur immunogénicité, les néoantigènes constituent des cibles émergentes pour l'immunothérapie du cancer, notamment les vaccins tumoraux, les thérapies cellulaires adoptives (ACT), les traitements à base d'anticorps et les prédicteurs potentiels pour les inhibiteurs de points de contrôle immunitaire (ICB) [3].
▲Caractéristiques moléculaires du gliome diffus de haut grade chez l'enfant[3]
Le nouvel antigène est composé de nouveaux antigènes personnalisés ciblant spécifiquement chaque patient ou de nouveaux antigènes communs exprimés chez de nombreux patients atteints de cancer. Les thérapies basées sur de nouveaux antigènes communs sont plus efficaces en termes de ressources et de temps que les thérapies basées sur de nouveaux antigènes personnalisés. Les nouveaux antigènes personnalisés étant spécifiques à chaque patient, ils ne peuvent pas être utilisés pour cibler un grand nombre de patients. Grâce aux dernières avancées en matière de séquençage à haut débit, les nouveaux antigènes personnalisés permettent au système immunitaire de cibler des épitopes immunogènes sur les tumeurs malignes sans antigènes communs prédéfinis.
Relation entre TMB et TNB
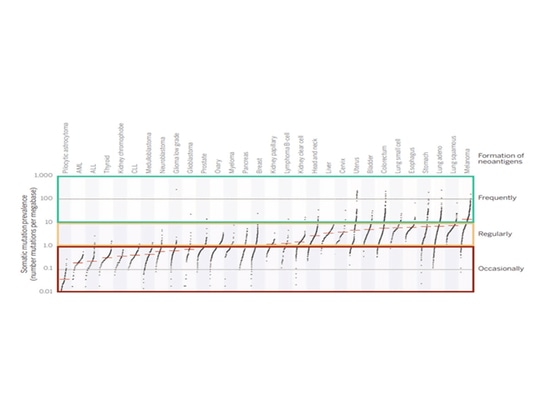
Chez la plupart des patients atteints de mélanome, le TMB est supérieur à 10, ce qui conduit à la génération de néoantigènes plus efficaces. Sur la base de ces données, on peut prédire que si le TMB est supérieur à 10 dans la tumeur, un nombre considérable de néoantigènes sera produit. Si le TMB est compris entre 1 et 10, il est encore possible de transporter des néoantigènes. En général, si le TMB est inférieur à 1 dans la tumeur, il est souvent difficile de générer des néoantigènes reconnaissables par les cellules T. Les tumeurs avec un TMB élevé (>10) indiquent qu'il y a plus de néoantigènes tumoraux à la surface des cellules tumorales, ce qui entraîne une destruction plus efficace par les cellules immunitaires. En outre, les patients ayant un TMB élevé peuvent présenter de meilleures réponses aux inhibiteurs de points de contrôle immunitaire.
▲Le TMB tumoral et le potentiel de production de néoantigènes[4]
Les défis de l'application clinique
1. L'immunothérapie basée sur de nouveaux antigènes ne montre une efficacité objective que dans un petit sous-ensemble de réponses de patients bien documentées. Par conséquent, des améliorations significatives sont nécessaires pour améliorer les résultats cliniques, notamment en augmentant la précision de la prédiction des nouveaux antigènes, en surmontant l'évasion immunitaire et en optimisant le pipeline pour le processus de production.
2. Précision limitée de la prédiction de nouveaux antigènes : L'application généralisée de l'immunothérapie personnalisée est limitée par la découverte restreinte de nouveaux antigènes spécifiques du cancer, en raison de l'hétérogénéité des charges mutationnelles et des différences significatives dans la présentation des nouveaux antigènes entre les différents types de tumeurs. Seulement 10 % des mutations non synonymes des cellules tumorales peuvent générer des peptides mutés ayant une forte affinité avec le CMH, et seulement 1 % des peptides liés au CMH sont reconnus par les cellules T du patient.
3. Perte de nouveaux antigènes : L'absence de nouveaux antigènes spécifiques de la tumeur peut constituer une stratégie d'évasion immunitaire cruciale pour les tumeurs. La perte de nouveaux antigènes peut être induite par différentes voies, telles que la perte du nombre de copies, la suppression transcriptionnelle, le silençage épigénétique et les mécanismes post-traductionnels.
4. Production insuffisante de nouvelles cellules T spécifiques de l'antigène : Le produit de détection génétique Panorama 602 de Foshu Bioscience comprend la détection de la perte d'hétérozygotie HLA-I pour évaluer les avantages de l'immunothérapie adjuvante. Il comprend également des informations sur les thérapies ciblées, la chimiothérapie, la charge mutationnelle tumorale (TMB), la charge néoantigénique tumorale (TNB), les facteurs immunitaires positifs et négatifs, ainsi que d'autres informations pertinentes sur l'utilisation des médicaments, le sous-typage, le pronostic, la génétique, etc.
Références
1. Nat Rev Cancer. 2017 Apr;17(4):209-222.
2.Viral Immunol. 2020 Apr;33(3):160-178.
3.Signal Transduct Target Ther. 2023 Jan 6;8(1):9.
4.Science. 2015 Apr 3;348(6230):69-74.
Déclaration : Cet article est destiné à être partagé. S'il existe des problèmes de droits d'auteur, veuillez nous contacter dès que possible et nous les corrigerons dans les plus brefs délais. Nous vous remercions de votre attention